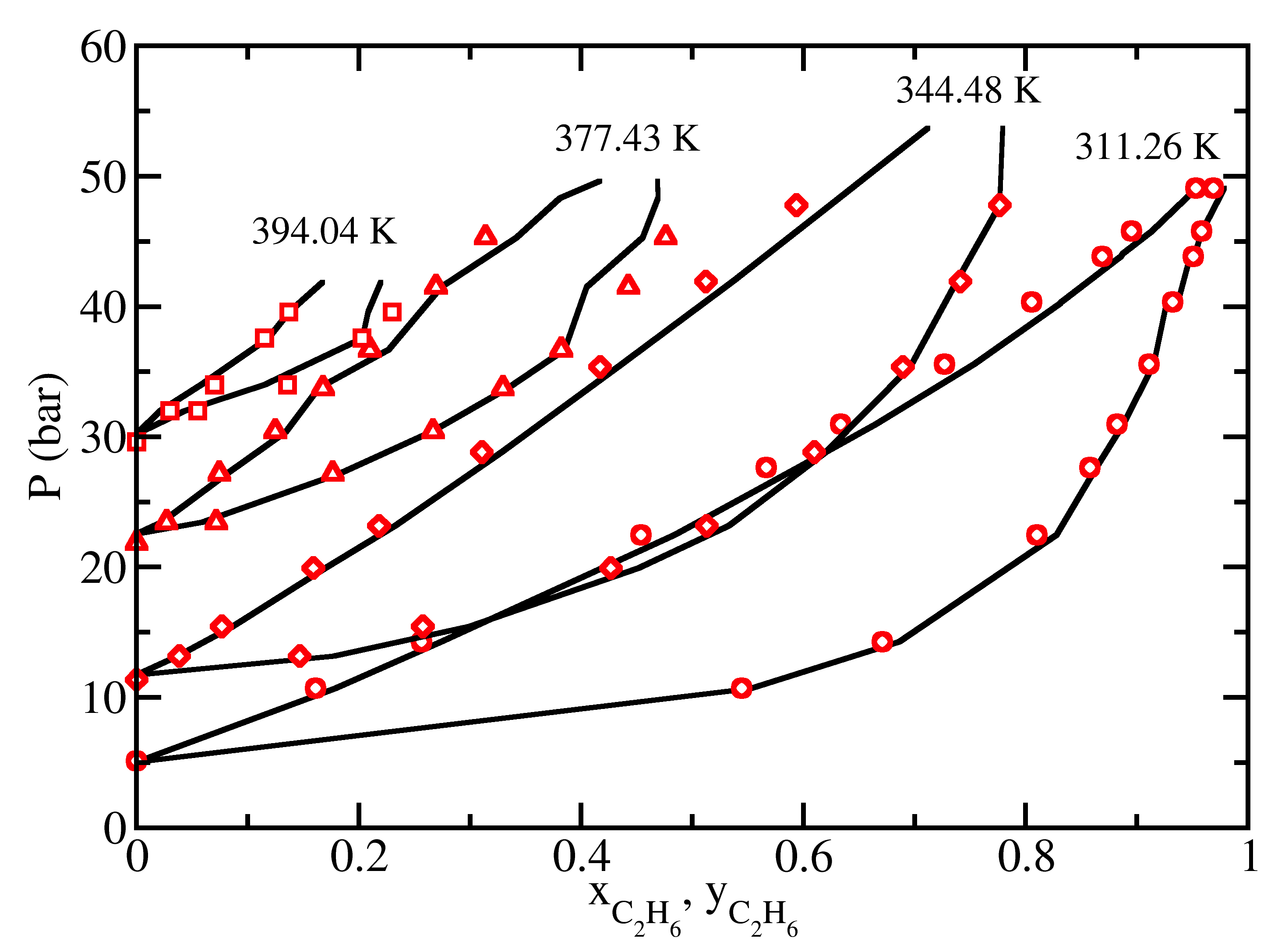
Vapor-Liquid Equilibrium
GOMC is capable of performing Gibbs ensemble Monte Carlo and grand canonical histogram-reweighting Monte Carlo simulations to predict the vapor-liquid equilibria of pure components and multicomponent mixtures. A variety of advanced configurational-bias algorithms, such as coupled-decoupled configurational-bias, molecular exchange Monte Carlo, configurational-bias regrowth, and crankshaft, are included to enhance the sampling of phase space.
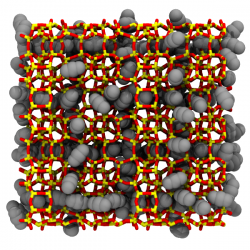
Adsorption
GOMC supports simulations of adsorption in rigid porous materials
and can be used for high through put screening of materials for
gas storage and separation. A tool to take structures from the
CoRE database
and automatically setup simulations for high
throughput screening may be found in our
GitHub repository.
In the future, our HTS code will be integrated into the Molecular
Simulation Design Framework (
MoSDeF
) toolkit.
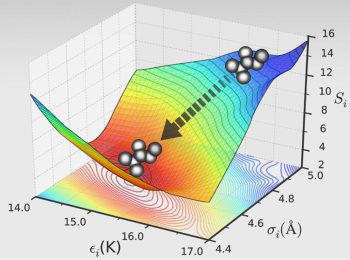
Force Field Optimization
GOMC has been widely used to optimize the non-bonded potential potential, using grand canonical histogram-reweighting Monte Carlo simulation. In addition, GOMC simulation output (simulation frame) can be combined with Multistate Bennett Acceptance Ratio ( MBAR ) to predict phase equilibria properties for an arbitrary force field parameter set that has not been simulated directly.
Gibbs Free Energy of Solvation
GOMC can predict Gibbs free energy of solvation in NPT Gibbs ensemble Monte Carlo simulation. Using the Molecular Exchange Monte Carlo move (MEMC) , the solute can be inserted into the dense system more efficiently. The ratio of number density between two phases at equilibrium, yield the Gibbs free energy of transfer.
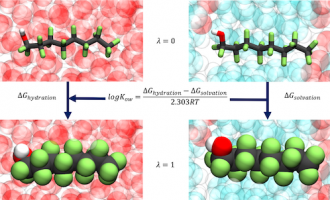
Free Energy Calculation
GOMC supports free energy calculation in NVT and NPT ensemble, using thermodynamic integration (TI) and free energy perturbation (FEP) methods, such as BAR and MBAR. Post analysis of GOMC energy output can be done using alchemlyb python library.
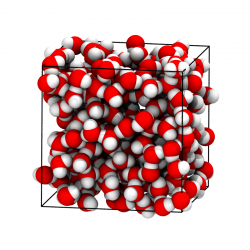
Condensed Phase Structure
GOMC can perform simulations in the NVT and NPT ensembles. GOMC outputs atomic coordinates in PDB and molecule connectivity in PSF format. This allows for straightforward visualization and analysis in VMD.
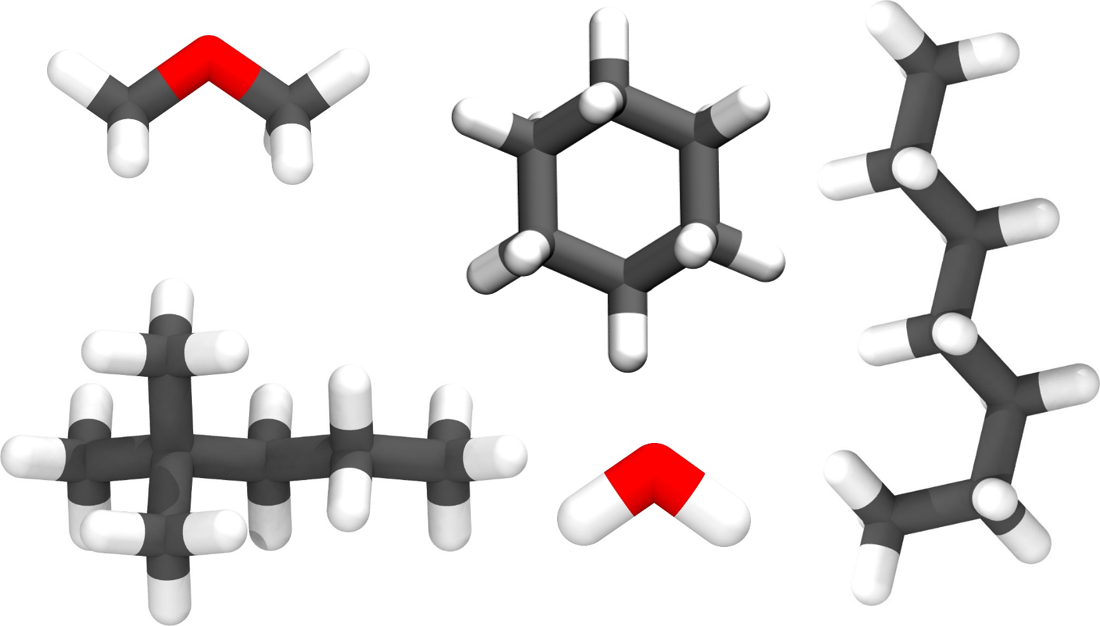
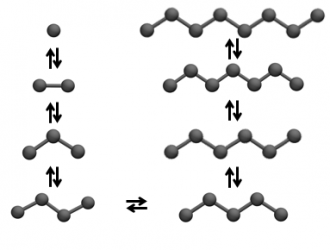
Support for a wide range of molecule topologies
With GOMC, one can simulate nearly any molecular topology, including multiple rings connected by flexible linkers.
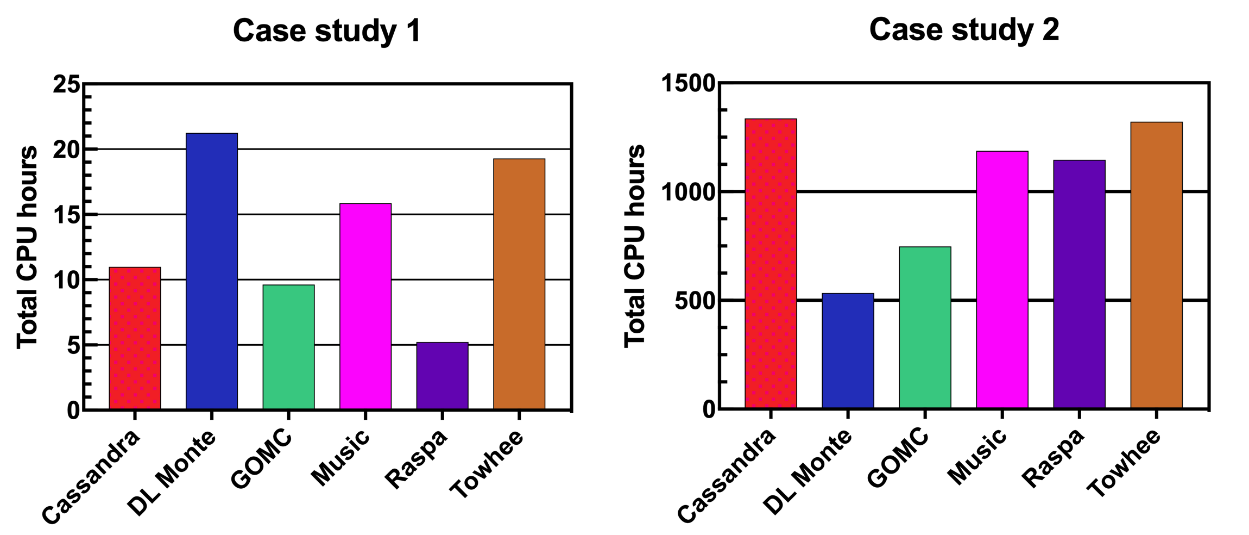
High Performance
GOMC exhibits both high sampling efficiency as well as excellent computational performance. GOMC has excellent single core performance, and also runs on multi-core CPUs using thread-level OpenMP parallelization, and on GPUs if available. Simulations of systems from ten to tens of thousands of atoms may be simulated efficiently with GOMC.