Input File Formats
In order to run simulation in GOMC, the following files need to be provided:
GOMC executable
PDB file(s)
PSF file(s)
Parameter file
Input file “NAME.conf” (proprietary control file)
In order to restart a simulation in GOMC from exactly where it left off, the following files also need to be provided:
XSC file(s)
COOR file(s)
CHK file
In order to run a hybrid MCMD simulation, the following files also need to be provided:
XSC file(s)
COOR file(s)
VEL files(s)
CHK file
PDB File
GOMC requires only one PDB file for NVT and NPT ensembles. However, GOMC requires two PDB files for GEMC and GCMC ensembles.
What is PDB file
The term PDB can refer to the Protein Data Bank (http://www.rcsb.org/pdb/), to a data file provided there, or to any file following the PDB format. Files in the PDB include various information such as the name of the compound, the ATOM and HETATM records containing the coordinates of the molecules, and etc. PDB widely used by NAMD, GROMACS, CHARMM, ACEMD, and Amber. GOMC ignore everything in a PDB file except for the REMARK, CRYST1, ATOM, and END records. An overview of the PDB standard can be found here:
http://www.wwpdb.org/documentation/file-format-content/format33/sect2.html#HEADER
http://www.wwpdb.org/documentation/file-format-content/format33/sect8.html#CRYST1
http://www.wwpdb.org/documentation/file-format-content/format33/sect9.html#ATOM
PDB contains four major parts; REMARK, CRYST1, ATOM, and END. Here is the definition of each field and how GOMC is using them to get the information it requires.
REMARK: This header records present experimental details, annotations, comments, and information not included in other records (for more information, click here).However, GOMC uses this header to print simulation informations.
Max Displacement (Å)
Max Rotation (Degree)
Max volume exchange (\(\AA^3\))
Monte Carlo Steps (MC)
CRYST1: This header records the unit cell dimension parameters.Lattice constant: a,b,c (Å)
Lattice angles: \(\alpha, \beta, \gamma\) (Degree)
ATOM: The ATOM records present the atomic coordinates for standard amino acids and nucleotides. They also present the occupancy and temperature factor for each atom.ATOM: Record name
serial: Atom serial number.
name: Atom name.
resName: Residue name.
chainID: Chain identifier.
resSeq: Residue sequence number.
x: Coordinates for X (Å).
y: Coordinates for Y (Å).
z: Coordinates for Z (Å).
occupancy: GOMC uses to define which atoms belong to which box.
beta: Beta or Temperature factor. GOMC uses this value to define the mobility of the atoms. element: Element symbol.
END: A frame in the PDB file is terminated with the keyword.
Here are the PDB output of GOMC for the first molecule of isobutane:
REMARK GOMC 122.790 3.14159 3439.817 1000000
CRYST1 35.245 35.245 35.245 90.00 90.00 90.00
ATOM 1 C1 ISB 1 0.911 -0.313 0.000 0.00 0.00 C
ATOM 2 C1 ISB 1 1.424 -1.765 0.000 0.00 0.00 C
ATOM 3 C1 ISB 1 -0.629 -0.313 0.000 0.00 0.00 C
ATOM 4 C1 ISB 1 1.424 0.413 -1.257 0.00 0.00 C
END
The fields seen here in order from left to right are the record type, atom ID, atom name, residue name, residue ID, x, y, and z coordinates, occupancy, temperature factor (called beta), and segment name.
The atom name is “C1” and residue name is “ISB”. The PSF file (next section) contains a lookup table of atoms. These contain the atom name from the PDB and the name of the atom kind in the parameter file it corresponds to. As multiple different atom names will all correspond to the same parameter, these can be viewed “atom aliases” of sorts. The chain letter (in this case ‘A’) is sometimes used when packing a number of PDBs into a single PDB file.
Important
VMD requires a constant number of ATOMs in a multi-frame PDB (multiple records terminated by “END” in a single file). To compensate for this, all atoms from all boxes in the system are written to the output PDBs of this code.
For atoms not currently in a box, the coordinates are set to
< 0.00, 0.00, 0.00 >. The occupancy is commonly just set to “1.00” and is left unused by many codes. We recycle this legacy parameter by using it to denote, in our output PDBs, the box a molecule is in (box 0 occupancy=0.00 ; box 1 occupancy=1.00)The beta value in GOMC code is used to define the mobility of the molecule.
Beta = 0.00: molecule can move and transfer within and between boxes.Beta = 1.00: molecule is fixed in its position.Beta = 2.00: molecule can move within the box but cannot be transferred between boxes.
Generating PDB file
With that overview of the format in mind, the following steps describe how a PDB file is typically built.
A single molecule PDB is obtained. In this example, the GaussView was used to draw the molecule, which was then edited by hand to adhere to the PDB spec properly. There are many open-source software that can build a molecule for you, such as Avagadro , molefacture in VMD and more. The end result is a PDB for a single molecule:
REMARK 1 File created by GaussView 5.0.8
ATOM 1 C1 ISB 1 0.911 -0.313 0.000 C
ATOM 2 C1 ISB 1 1.424 -1.765 0.000 C
ATOM 3 C1 ISB 1 -0.629 -0.313 0.000 C
ATOM 4 C1 ISB 1 1.424 0.413 -1.257 C
END
Next, packings are calculated to place the simulation in a region of vapor-liquid coexistence. There are a couple of ways to do this in Gibbs ensemble:
Pack both boxes to a single middle density, which is an average of the liquid and vapor densities.
Same as previous method, but add a modest amount to axis of one box (e.g. 10-30 A). This technique can be handy in the constant pressure Gibbs ensemble.
Pack one box to the predicted liquid density and the other to the vapor density.
A good reference for getting the information needed to estimate packing is the NIST Web Book database of pure compounds:
After packing is determined, a basic pack can be performed with a Packmol script. Here is the example of packing 1000 isobutane in 70 A cubic box:
tolerance 3.0
filetype pdb
output STEP2_ISB_packed_BOX 0.pdb
structure isobutane.pdb
number 1000
inside cube 0.1 0.1 0.1 70.20
end structure
Copy the above text into “pack_isobutane.inp” file, save it and run the script by typing the following line into the terminal:
$ ./packmol < pack_isobutane.inp
PSF File
GOMC requires only one PSF file for NVT and NPT ensembles. However, GOMC requires two PSF files for GEMC and GCMC ensembles.
What is PSF file
Protein structure file (PSF), contains all of the molecule-specific information needed to apply a particular force field to a molecular system. The CHARMM force field is divided into a topology file, which is needed to generate the PSF file, and a parameter file, which supplies specific numerical values for the generic CHARMM potential function. The topology file defines the atom types used in the force field; the atom names, types, bonds, and partial charges of each residue type; and any patches necessary to link or otherwise mutate these basic residues. The parameter file provides a mapping between bonded and nonbonded interactions involving the various combinations of atom types found in the topology file and specific spring constants and similar parameters for all of the bond, angle, dihedral, improper, and van der Waals terms in the CHARMM potential function. PSF file widely used by by NAMD, CHARMM, and X-PLOR.
The PSF file contains six main sections: remarks, atoms, bonds, angles, dihedrals, and impropers (dihedral force terms used to maintain
planarity). Each section starts with a specific header described bellow:
NTITLE: remarks on the file. The following is taken from a PSF file for isobutane:PSF 3 !NTITLE REMARKS original generated structure x-plor psf file REMARKS topology ./Top_Branched_Alkanes.inp REMARKS segment ISB { first NONE; last NONE; auto angles dihedrals }NATOM: Defines the atom names, types, and partial charges of each residue type.atom ID segment name residue ID residue name atom name atom type atom charge atom mass
The following is taken from a PSF file for isobutane:
4000 !NATOM 1 ISB 1 ISB C1 CH1 0.000000 13.0190 0 2 ISB 1 ISB C2 CH3 0.000000 15.0350 0 3 ISB 1 ISB C3 CH3 0.000000 15.0350 0 4 ISB 1 ISB C4 CH3 0.000000 15.0350 0 5 ISB 2 ISB C1 CH1 0.000000 13.0190 0 6 ISB 2 ISB C2 CH3 0.000000 15.0350 0 7 ISB 2 ISB C3 CH3 0.000000 15.0350 0 8 ISB 2 ISB C4 CH3 0.000000 15.0350 0
The fields in the atom section, from left to right are atom ID, segment name, residue ID, residue name, atom name, atom type, charge, mass, and an unused 0.
NBOND: The covalent bond section lists four pairs of atoms per line. The following is taken from a PSF file for isobutane:3000 !BOND: bonds 1 2 1 3 1 4 5 6 5 7 5 8
NTHETA: The angle section lists three triples of atoms per line. The following is taken from a PSF file for isobutane:3000 !NTHETA: angles 2 1 4 2 1 3 3 1 4 6 5 8 6 5 7 7 5 8
NPHI: The dihedral sections list two quadruples of atoms per line.NIMPHI: The improper sections list two quadruples of atoms per line. GOMC currently does not support improper. For the molecules without dihedral or improper, PDF file look like the following:0 !NPHI: dihedrals 0 !NIMPHI: impropers
(other sections such as cross terms)
Important
The PSF file format is a highly redundant file format. It repeats identical topology of thousands of molecules of a common kind in some cases. GOMC follows the same approach as NAMD, allowing this excess information externally and compiling it in the code.
Other sections (e.g. cross terms) contain unsupported or legacy parameters and are ignored.
Following the restriction of VMD, the order of the atoms in PSF file must match the order of the atoms in the PDB file.
Improper entries are read and stored, but are not currently used. Support will eventually be added for this.
Generating PSF file
The PSF file is typically generated using PSFGen. It is convenient to make a script, such as the example below, to do this:
package require psfgen
topology ./Top_branched_Alaknes.inp
segment ISB {
pdb ./STEP2_ISB_packed_BOX 0.pdb
first none
last none
}
coordpdb ./STEP2_ISB_packed_BOX 0.pdb ISB
writepsf ./STEP3_START_ISB_sys_BOX_0.psf
writepdb ./STEP3_START_ISB_sys_BOX_0.pdb
Typically, one script is run per box to generate a finalized PDB/PSF for that box. The script requires one additional file, the NAMD-style topology file. While GOMC does not directly read or interact with this file, it’s typically used to generate the PSF and, hence, is considered one of the integral file types. It will be briefly discussed in the following section.
Topology File
A CHARMM forcefield topology file contains all of the information needed to convert a list of residue names into a complete PSF structure file. The topology is a whitespace separated file format, which contains a list of atoms and their corresponding masses, and a list of residue information (charges, composition, and topology). Essentially, it is a non-redundant lookup table equivalent to the PSF file.
This is followed by a series of residues, which tell PSFGen what atoms are bonded to a given atom. Each residue is comprised of four key elements:
A header beginning with the keyword RESI with the residue name and net charge
A body with multiple ATOM entries (not to be confused with the PDB-style entries of the same name), which list the partial charge on the particle and what kind of atom each named atom in a specific molecule/residue is.
A section of lines starting with the word BOND contains pairs of bonded atoms (typically 3 per line)
A closing section with instructions for PSFGen.
Here’s an example of topology file for isobutane:
* Custom top file -- branched alkanes *
11
!
MASS 1 CH3 15.035 C !
MASS 2 CH1 13.019 C !
AUTOGENERATE ANGLES DIHEDRALS
RESI ISB 0.00 ! isobutane - TraPPE
GROUP
ATOM C1 CH1 0.00 ! C3\
ATOM C2 CH3 0.00 ! C1-C2
ATOM C3 CH3 0.00 ! C4/
ATOM C4 CH3 0.00 !
BOND C1 C2 C1 C3 C1 C4
PATCHING FIRS NONE LAST NONE
END
Note
The keyword END must be used to terminate this file and keywords related to the auto-generation process must be placed near the top of the file, after the MASS definitions.
Tip
More in-depth information can be found in the following links:
Parameter File
Currently, GOMC uses a single parameter file and the user has the two kinds of parameter file choices:
CHARMM(Chemistry at Harvard Molecular Mechanics) compatible parameter fileEXOTICorMieparameter file
If the parameter file type is not specified or if the chosen file is missing, an error will result.
Both force field file options are whitespace separated files with sections preceded by a tag. When a known tag (representing a molecular interaction in the model) is encountered, reading of that section of the force field begins. Comments (anything after a * or !) and whitespace are ignored. Reading concludes when the end of the file is reached or another section tag is encountered.
CHARMM format parameter file
CHARMM contains a widely used model for describing energies in Monte Carlo and molecular dynamics simulations. It is intended to be compatible with other codes that use such a format, such as NAMD. See here for a general overview of the CHARMM force field.
Here’s the basic CHARMM contributions that are supported in GOMC:
As seen above, the following are recognized, read and used:
BONDS- Quadratic expression describing bond stretching based on bond length (b) in Angstrom – Typically, it is ignored as bonds are rigid for Monte Carlo simulations.Note
GOMC does not sample bond stretch. To ignore the relative bond energy, set the \(K_b\) to a large value i.e. “999999999999”.
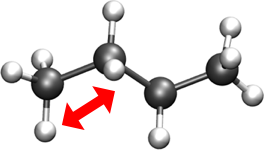
Oscillations about the equilibrium bond length
ANGLES- Describe the conformational behavior of an angle (\(\delta\)) between three atoms, one of which is shared branch point to the other two.Note
To fix any angle and ignore the related angle energy, set the \(K_\theta\) to a large value i.e. “999999999999”.
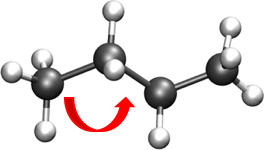
Oscillations of 3 atoms about an equilibrium bond angle
DIHEDRALS- Describes crankshaft-like rotation behavior about a central bond in a series of three consecutive bonds (rotation is given as \(\phi\)).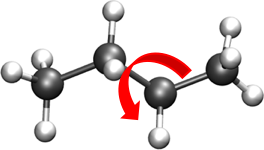
Torsional rotation of 4 atoms about a central bond
NONBONDED- This tag name only should be used if CHARMM force files are being used. This section describes 12-6 (Lennard-Jones) non-bonded interactions. Non-bonded parameters are assigned by specifying atom type name followed by polarizabilities (which will be ignored), minimum energy, and (minimum radius)/2. In order to modify 1-4 interaction, a second polarizability (again, will be ignored), minimum energy, and (minimum radius)/2 need to be defined; otherwise, the same parameter will be considered for 1-4 interaction.
Non-bonded energy terms (electrostatics and Lennard-Jones)
NBFIX- This tag name only should be used if CHARMM force field is being used. This section allows in- teraction between two pairs of atoms to be modified, done by specifying two atom type names followed by minimum energy and minimum radius. In order to modify 1-4 interaction, a second minimum energy and minimum radius need to be defined; otherwise, the same parameter will be considered for 1-4 interaction.Note
Please pay attention that in this section we define minimum radius, not (minimum radius)/2 as it is defined in the NONBONDED section.
Note
This does not modify the 1-4 electrostatic interactions.
Currently, supported sections of the
CHARMMcompliant file includeBONDS,ANGLES,DIHEDRALS,NONBONDED,NBFIX. Other sections such asCMAPare not currently read or supported.
BONDS
(“bond stretching”) is one key section of the CHARMM-compliant file. Units for the \(K_b\) variable in this section are in kcal/mol; the \(b_0\) section (which represents the equilibrium bond length for that kind of pair) is measured in Angstroms.
BONDS
!V(bond) = Kb(b - b0)**2
!
!Kb: kcal/mole/A**2
!b0: A
!
!Kb (kcal/mol) = Kb (K) * Boltz. const.;
!
!atom type Kb b0 description
CH3 CH1 9999999999 1.540 ! TraPPE 2
Note
The \(K_b\) value may appear odd, but this is because a larger value corresponds to a more rigid bond. As Monte Carlo force fields (e.g. TraPPE) typically treat molecules as rigid constructs, \(K_b\) is set to a large value - 9999999999. Sampling bond stretch is not supported in GOMC.
ANGLES
(“bond bending”), where \(\theta\) is the measured bond angle and \(\theta_0\) is the equilibrium bond angle for that kind of pair, are commonly measured in degrees and \(K_\theta\) is the force constant measured in kcal/mol/K. These values, in literature, are often expressed in Kelvin (K).
To convert Kelvin to kcal/mol/K, multiply by the Boltzmann constant – \(K_\theta\), 0.0019872041 kcal/mol. In order to fix the angle, it requires to set a large value for \(K_\theta\). By assigning a large value like 9999999999, specified angle will be fixed and energy of that angle will considered to be zero.
Here is an example of what is necessary for isobutane:
ANGLES
!
!V(angle) = Ktheta(Theta - Theta0)**2
!
!V(Urey-Bradley) = Kub(S - S0)**2
!
!Ktheta: kcal/mole/rad**2
!Theta0: degrees
!S0: A
!
!Ktheta (kcal/mol) = Ktheta (K) * Boltz. const.
!
!atom types Ktheta Theta0
CH3 CH1 CH3 62.100125 112.00 ! TraPPE 2
Some CHARMM ANGLES section entries include Urey-Bradley potentials (\(K_{ub}\), \(b_{ub}\)), in addition to the standard quadratic angle potential. The constants related to this potential function are currently read, but the logic has not been added to calculate this potential function. Support for this potential function will be added in later versions of the code.
DIHEDRALS
The final major bonded interactions section of the CHARMM compliant parameter file are the DIHEDRALS. Dihedral energies were represented by a cosine series where \(\phi\) is the dihedral angle, \(C_n\) are dihedral force constants, \(n\) is the multiplicity, and \(\delta_n\) is the phase shift. Often, there are 4 to 6 terms in a dihedral. Angles for the dihedrals’ deltas are given in degrees.
Since isobutane has no dihedral, here are the parameters pertaining to 2,3-dimethylbutane:
DIHEDRALS
!
!V(dihedral) = Kchi(1 + cos(n(chi) - delta))
!
!Kchi: kcal/mole
!n: multiplicity
!delta: degrees
!
!Kchi (kcal/mol) = Kchi (K) * Boltz. const.
!
!atom types Kchi n delta description
X CH1 CH1 X -0.498907 0 0.0 ! TraPPE 2
X CH1 CH1 X 0.851974 1 0.0 ! TraPPE 2
X CH1 CH1 X -0.222269 2 180.0 ! TraPPE 2
X CH1 CH1 X 0.876894 3 0.0 ! TraPPE 2
Note
The code allows the use of ‘X’ to indicate ambiguous positions on the ends. This is useful because this kind is often determined solely by the two middle atoms in the middle of the dihedral, according to literature.
Note
If a dihedral parameter was defined with multiplicity value of zero (\(n\) = 0), GOMC will automatically assign the phase shift value to 90 (\(\delta_n\) = 90) to recover the above dihedral expresion.
IMPROPERS
Energy parameters used to describe out-of-plane rocking are currently read, but unused. The section is often blank. If it becomes necessary, algorithms to calculate the improper energy will need to be added.
NONBONDED
The next section of the CHARMM style parameter file is the NONBONDED. The nonbonded energy in CHARMM is presented as 12-6 potential where, \(r_{ij}\), \(\epsilon_{ij}\), \({R_{min}}_{ij}\) are the separation, minimum potential, and minimum potential distance, respectively. In order to use TraPPE this section of the CHARMM compliant file is critical.
Here’s an example with our isobutane potential model:
NONBONDED
!
!V(Lennard-Jones) = Eps,i,j[(Rmin,i,j/ri,j)**12 - 2(Rmin,i,j/ri,j)**6]
!
!atom ignored epsilon Rmin/2 ignored eps,1-4 Rmin/2,1-4
CH3 0.0 -0.194745992 2.10461634058 0.0 0.0 0.0 ! TraPPE 1
CH1 0.0 -0.019872040 2.62656119304 0.0 0.0 0.0 ! TraPPE 2
End
Note
The \(R_{min}\) is the potential well-depth, where the attraction is maximum. However, \(\sigma\) is the particle diameter, where the interaction energy is zero. To convert \(\sigma\) to \(R_{min}\), simply multiply \(\sigma\) by 0.56123102415.
Important
If no parameter was defined for 1-4 interaction e.g (\(\epsilon_{1-4}, Rmin_{1-4}/2\)), GOMC will use the \(\epsilon, Rmin/2\) for 1-4 interaction.
NBFIX
The last section of the CHARMM style parameter file is the NBFIX. In this section, individual pair interaction will be modified. First, pseudo non-bonded parameters have to be defined in NONBONDED and modified in NBFIX. Here iss an example if it is required to modify interaction between CH3 and CH1 atoms:
NBFIX
!V(Lennard-Jones) = Eps,i,j[(Rmin,i,j/ri,j)**12 - 2(Rmin,i,j/ri,j)**6]
!
!atom atom epsilon Rmin eps,1-4 Rmin,1-4
CH3 CH1 -0.294745992 1.10461634058 !
End
Important
If no parameter was defined for 1-4 interaction e.g (\(\epsilon_{1-4}, Rmin_{1-4}\)), GOMC will use the \(\epsilon, Rmin\) for 1-4 interaction.
Exotic or Mie Parameter File
The Mie file is intended for use with nonstandard/specialty models of molecular interaction, which are not included in CHARMM standard.
Mie Potential
where \(r_{ij}\), \(\epsilon_{ij}\), and \(\sigma_{ij}\) are, respectively, the separation, minimum potential, and collision diameter for the pair of interaction sites \(i\) and \(j\). The constant \(C_n\) is a normalization factor such that the minimum of the potential remains at \(-\epsilon_{ij}\) for all \(n_{ij}\). In the 12-6 potential, \(C_n\) reduces to the familiar value of 4.
Buckingham Potential (Exp-6)
where \(r_{ij}\), \(\epsilon_{ij}\), and \(R_{min,ij}\) are, respectively, the separation, minimum potential, and minimum potential distance for the pair of interaction sites \(i\) and \(j\). The constant \(\alpha_{ij}\) is an exponential-6 parameter. The cutoff distance \(R_{max,ij}\) is the smallest positive value for which \(\frac{dE_{ij}}{dr_{ij}}=0\).
Note
In order to use Mie or Exotice potential file format for Buckingham potential, instead of defining \(R_{min}\), we define \(\sigma\) (collision diameter or the distance, where potential is zero)
and GOMC will calculate the \(R_{min}\) and \(R_{max}\) using Buckingham potential equation.
Currently, two custom interaction are included:
NONBODED_MIEThis section describes n-6 (Lennard-Jones) or Exp-6 (Buckingham) non-bonded interactions. The Lennard-Jones potential (12-6) is a subset of Mie potential. Non-bonded parameters are assigned by specifying the following fields in order:Atom type name
Minimum energy (\(\epsilon\))
Atom diameter (\(\sigma\))
Repulsion exponent (\(n\)) in
Miepotential or \(\alpha\) inBuckinghampotential.
The 1-4 interaction can be modified by specifying the following fields in order:
Minimum energy (\(\epsilon_{1-4}\))
Atom diameter (\(\sigma_{1-4}\))
Repulsion exponent (\(n_{1-4}\)) in
Miepotential or \(\alpha_{1-4}\) inBuckinghampotential.
Note
If no parameter is provided for 1-4 interaction, same parameters (item 2, 3, 4) would be considered for 1-4 interaction.
NBFIX_MIEThis section allows n-6 (Lennard-Jones) or Exp-6 (Buckingham) interaction between two pairs of atoms to be modified. Interaction between two pairs of atoms can be modified by specifying the following fields in order:Atom type 1 name
Atom type 2 name
Minimum energy (\(\epsilon\))
Atom diameter (\(\sigma\))
Repulsion exponent (\(n\)) in
Miepotential or \(\alpha\) inBuckinghampotential.
The 1-4 interaction between two pairs of atoms can be modified by specifying the following fields in order:
Minimum energy (\(\epsilon_{1-4}\))
Atom diameter (\(\sigma_{1-4}\))
Repulsion exponent (\(n_{1-4}\)) in
Miepotential or \(\alpha_{1-4}\) inBuckinghampotential.
Note
If no parameter is provided for 1-4 interaction, same parameters (item 3, 4, 5) would be considered for 1-4 interaction.
Note
In Mie or Buckingham potential, the definition of atom diameter(\(\sigma\)) is same for both NONBONDED_MIE and NBFIX_MIE.
Important
If no parameter was defined for 1-4 interaction e.g (\(\epsilon_{1-4}, \sigma_{1-4}, n_{1-4}\)), GOMC will use the \(\epsilon, \sigma, n\) for 1-4 interaction.
Otherwise, the Mie file reuses the same geometry section headings - BONDS / ANGLES / DIHEDRALS / etc. The only difference in these sections versus in the CHARMM format force field file is that the energies are in Kelvin (‘K’), the unit most commonly found for parameters in Monte Carlo chemical simulation literature. This precludes the need to convert to kcal/mol, the energy unit used in CHARMM. The most frequently used section of the Mie files in the Mie potential section is NONBONDED_MIE.
Here is the example of Mie or Exotic parameters file format that are used to simulate alkanes with Mie potential:
NONBONDED_MIE
!
!V(Mie) = const*eps*((sig/r)^n-(sig/r)^6)
!
!atom eps sig n eps,1-4 sig,1-4 n,1-4
CH4 161.00 3.740 14 0.0 0.0 0.0 ! Potoff, et al. '09
CH3 121.25 3.783 16 0.0 0.0 0.0 ! Potoff, et al. '09
CH2 61.00 3.990 16 0.0 0.0 0.0 ! Potoff, et al. '09
NBFIX_MIE
!V(Mie) = const*eps*((sig/r)^n-(sig/r)^6)
!
!atom atom epsilon sig n eps,1-4 sig,1-4 n,1-4
CH3 CH2 100.00 3.8 16 0.0 0.0 0.0 !
End
Here is the example of Mie or Exotic parameters file format that are used to simulate water with Buckingham potential:
NONBONDED_MIE
!
!V(exp-6) = ((eps-ij * alpha)/(alpha - 6)) * ((6 / alpha) * exp(alpha * [1 - (r / rmin)]) - (rmin / r)^6))
!
!atom eps sig alpha eps,1-4 sig,1-4 n,1-4
OT 159.78 3.195 12 0.0 0.0 0.0 ! Errington, et al. 1998
HT 0.0 0.0 0 0.0 0.0 0.0 ! Errington, et al. 1998
NBFIX_MIE
!V(exp-6) = ((eps-ij * alpha)/(alpha - 6)) * ((6 / alpha) * exp(alpha * [1 - (r / rmin)]) - (rmin / r)^6))
!
!atom atom epsilon sig alpha eps,1-4 sig,1-4 n,1-4
HT OT 0.00 0.0 0 0.0 0.0 0.0 !
End
Note
Although the units (Angstroms) are the same, the Mie file uses \(\sigma\), not the \(R_{min}\) used by CHARMM. The energy in the exotic file are expressed in Kelvin (K), as this is the standard convention in the literature.
Control File (*.conf)
The control file is GOMC’s proprietary input file. It contains key settings. The settings generally fall under three categories:
Input/Simulation Setup
System Settings for During Run
Output Settings
Note
The control file is designed to recognize logic values, such as “yes/true/on” or “no/false/off”. The keyword in control file is not case sensitive.
Input/Simulation Setup
In this section, input file names are listed. In addition, if you want to restart your simulation or use integer seed for running your simulation, you need to modify this section according to your purpose.
RestartDetermines whether to restart the simulation from restart file (*_restart.pdb) or not.
Value 1: Boolean - True if restart, false otherwise.
ExpertModeDetermines whether to perform error checking of move selection to ensure correct ensemble is sampled. This allows the user to run a simulation with no volume moves in NPT, NPT-GEMC; no molecule transfers in GCMC, GEMC.
Value 1: Boolean - True if enable expert mode; false otherwise.
CheckpointDetermines whether to restart the simulation from checkpoint file or not. Restarting the simulation with would result in an identitcal outcome, as if previous simulation was continued. This is required for hybrid Monte-Carlo Molecular Dyanamics in open-ensembles (GCMC/GEMC) to concatenate trajectory files since the molecular transfers rearranges the order of the molecules. Checkpointing will ensure the molecules are loaded in the same order each cycle.
Value 1: Boolean - True if restart with checkpoint file, false otherwise.
Value 2: String - Sets the name of the checkpoint file.
Checkpoint true AR_KR_continued.chk
PRNGDictates how to start the pseudo-random number generator (PRNG)
Value 1: String
RANDOM: Randomizes Mersenne Twister PRNG with random bits based on the system time.
################################# # kind {RANDOM, INTSEED} ################################# PRNG RANDOMINTSEED: This option “seeds” the Mersenne Twister PRNG with a standard integer. When the same integer is used, the generated PRNG stream should be the same every time, which is helpful in tracking down bugs.
Random_SeedDefines the seed number. If “INTSEED” is chosen, seed number needs to be specified; otherwise, the program will terminate.
Value 1: ULONG - If “INTSEED” option is selected for PRNG (See bellow example)
################################# # kind {RANDOM, INTSEED} ################################# PRNG INTSEED Random_Seed 50ParaTypeCHARMMSets force field type to CHARMM style.
Value 1: Boolean - True if it is CHARMM forcefield, false otherwise.
################################# # FORCE FIELD TYPE ################################# ParaTypeCHARMM true
ParaTypeEXOTICorParaTypeMieSets force field type to Mie style.
Value 1: Boolean - True if it is Mie forcefield, false otherwise.
################################# # FORCE FIELD TYPE ################################# ParaTypeEXOTIC true
ParaTypeMARTINISets force field type to MARTINI style.
Value 1: Boolean - True if it is MARTINI forcefield, false otherwise.
################################# # FORCE FIELD TYPE ################################# ParaTypeMARTINI true
ParametersProvides the name and location of the parameter file to use for the simulation.
Value 1: String - Sets the name of the parameter file.
################################# # FORCE FIELD TYPE ################################# ParaTypeCHARMM yes Parameters ../../common/Par_TraPPE_Alkanes.inp Parameters ../../common/Par_TIP4P.inp
Note
More than one parameter file can be provided.
CoordinatesDefines the PDB file names (coordinates) and location for each box in the system.
Value 1: Integer - Sets box number (starts from ‘0’).
Value 2: String - Sets the name of PDB file.
Note
NVT and NPT ensembles requires only one PDB file and GEMC/GCMC requires two PDB files. If the number of PDB files is not compatible with the simulation type, the program will terminate.
Example of NVT or NPT ensemble:
############################################# # INPUT PDB FILES - NVT or NPT ensemble ############################################# Coordinates 0 STEP3_START_ISB_sys.pdb
Example of Gibbs or GC ensemble:
############################################# # INPUT PDB FILES - Gibbs or GCMC ensemble ############################################# Coordinates 0 STEP3_START_ISB_sys_BOX_0.pdb Coordinates 1 STEP3_START_ISB_sys_BOX_1.pdb
Note
In case of
Restarttrue, the restart PDB output file from GOMC (OutputName_BOX_N_restart.pdb) can be used for each box.Example of Gibbs ensemble when Restart mode is active:
################################# # INPUT PDB FILES ################################# Coordinates 0 ISB_T_270_k_BOX_0_restart.pdb Coordinates 1 ISB_T_270_k_BOX_1_restart.pdb
StructuresDefines the PSF filenames (structures) for each box in the system.
Value 1: Integer - Sets box number (start from ‘0’)
Value 2: String - Sets the name of PSF file.
Note
NVT and NPT ensembles requires only one PSF file and GEMC/GCMC requires two PSF files. If the number of PSF files is not compatible with the simulation type, the program will terminate.
Example of NVT or NPT ensemble:
################################# # INPUT PSF FILES ################################# Structure 0 STEP3_START_ISB_sys.psf
Example of Gibbs or GC ensemble:
################################# # INPUT PSF FILES ################################# Structure 0 STEP3_START_ISB_sys_BOX_0.psf Structure 1 STEP3_START_ISB_sys_BOX_1.psf
Note
In case of
Restarttrue, the PSF output file from GOMC (OutputName_BOX_N_restart.psf) can be used for both boxes.Example of Gibbs ensemble when
Restartmode is active:################################# # INPUT PSF FILES ################################# Structure 0 ISB_T_270_k_BOX_0_restart.psf Structure 1 ISB_T_270_k_BOX_1_restart.psf
binCoordinatesDefines the DCD file names (coordinates) and location for each box in the system.
Value 1: Integer - Sets box number (starts from ‘0’).
Value 2: String - Sets the name of PDB file.
Note
NVT and NPT ensembles requires only one DCD file and GEMC/GCMC requires only one PDB files, although loading two is supported. This is different from PDB files, for which two are required in GEMC/GCMC. This allows the user to only load binary coordinates for one box.
Example of NVT or NPT ensemble:
############################################# # INPUT PDB FILES - NVT or NPT ensemble ############################################# binCoordinates 0 STEP3_START_ISB_sys.coor
Example of Gibbs or GC ensemble:
############################################# # INPUT PDB FILES - Gibbs or GCMC ensemble ############################################# binCoordinates 0 STEP3_START_ISB_sys_BOX_0.coor binCoordinates 1 STEP3_START_ISB_sys_BOX_1.coor
Note
In case of
Restart, the restart DCD output file from GOMC (OutputName_BOX_N_restart.coor) can be used for each box.Example of Gibbs ensemble when Restart mode is active:
################################# # INPUT PDB FILES ################################# binCoordinates 0 ISB_T_270_k_BOX_0_restart.coor binCoordinates 1 ISB_T_270_k_BOX_1_restart.coor
binVelocitiesDefines the VEL file names (velocities) and location for each box in the system.
Value 1: Integer - Sets box number (starts from ‘0’).
Value 2: String - Sets the name of VEL file.
Note
Originate from a Molecular Dynamics softwrae such as NAMD. GOMC will only output a velocity restart file if it is provided one using this keyword.
Note
In hybrid Monte-Carlo Molecular Dynamics, the velocities of the atoms should be preserved across cycles to increase accuracy. These files are not used internally by GOMC, only maintained. If a molecular transfer occurs, a new velocity is generated by Langevin dynamics.
Example of NVT or NPT ensemble:
############################################# # INPUT PDB FILES - NVT or NPT ensemble ############################################# binVelocities 0 STEP3_START_ISB_sys.vel
Example of Gibbs or GC ensemble:
############################################# # INPUT PDB FILES - Gibbs or GCMC ensemble ############################################# binVelocities 0 STEP3_START_ISB_sys_BOX_0.vel binVelocities 1 STEP3_START_ISB_sys_BOX_1.vel
Note
In case of
Restart, the restart VEL output file from GOMC (OutputName_BOX_N_restart.vel) can be used for each box.Example of Gibbs ensemble when Restart mode is active:
################################# # INPUT PDB FILES ################################# binVelocities 0 ISB_T_270_k_BOX_0_restart.vel binVelocities 1 ISB_T_270_k_BOX_1_restart.vel
extendedSystemDefines the XSC file names (box dimensions and origin) and location for each box in the system.
Value 1: Integer - Sets box number (starts from ‘0’).
Value 2: String - Sets the name of XSC file.
Note
Previously, this information was stored in plain-text format at the top of restart PDB files. This will be deprecated in favor of binary XSC.
Example of NVT or NPT ensemble:
############################################# # INPUT PDB FILES - NVT or NPT ensemble ############################################# extendedSystem 0 STEP3_START_ISB_sys.xsc
Example of Gibbs or GC ensemble:
############################################# # INPUT PDB FILES - Gibbs or GCMC ensemble ############################################# extendedSystem 0 STEP3_START_ISB_sys_BOX_0.xsc extendedSystem 1 STEP3_START_ISB_sys_BOX_1.xsc
Note
In case of
Restart, the restart XSC output file from GOMC (OutputName_BOX_N_restart.xsc) can be used for each box.Example of Gibbs ensemble when Restart mode is active:
################################# # INPUT PDB FILES ################################# extendedSystem 0 ISB_T_270_k_BOX_0_restart.xsc extendedSystem 1 ISB_T_270_k_BOX_1_restart.xsc
MultiSimFolderNameThe name of the folder to be created which contains output from the multisim.
Value 1: String - Name of the folder to contain output
MultiSimFolderName outputFolderName
System Settings for During Run Setup
This section contains all the variables not involved in the output of data during the simulation, or in the reading of input files at the start of the simulation. In other words, it contains settings related to the moves, the thermodynamic constants (based on choice of ensemble), and the length of the simulation. Note that some tags, or entries for tags, are only used in certain ensembles (e.g. Gibbs ensemble). These cases are denoted with colored text.
GEMC(For Gibbs Ensemble runs only) Defines the type of Gibbs Ensemble simulation you want to run. If neglected in Gibbs Ensemble, it simply defaults to const volume (NVT) Gibbs Ensemble.
Value 1: String - Allows you to pick between isovolumetric (“NVT”) and isobaric (“NPT”) Gibbs ensemble simulations.
Note
The default value for
GEMCis NVT.################################# # GEMC TYPE (DEFAULT IS NVT GEMC) ################################# GEMC NVT
PressureFor
NPTorNPT-GEMCsimulation, imposed pressure (in bar) needs to be specified; otherwise, the program will terminate.Value 1: Double - Constant pressure in bar.
################################# # GEMC TYPE (DEFAULT IS NVT GEMC) ################################# GEMC NPT Pressure 5.76
TemperatureSets the temperature at which the system will run.
Value 1: Double - Constant temperature of simulation in degrees Kelvin.
################################# # SIMULATION CONDITION ################################# Temperature 270.00
(MPI-GOMC Only)
Value 1: List of Doubles - A list of constant temperatures for simulations in degrees Kelvin.
################################# # SIMULATION CONDITION ################################# Temperature 270.00 280.00 290.00 300.00
Note
To use more than one temperature, GOMC must be compiled in MPI mode. Also, if GOMC is compiled in MPI mode, more than one temperature is required. To use only one temperature, use standard GOMC.
RcutSets a specific radius that non-bonded interaction energy and force will be considered and calculated using defined potential function.
Value 1: Double - The distance to truncate the Lennard-Jones potential at.
RcutLowSets a specific minimum possible in angstrom that reject any move that places any atom closer than specified distance.
Value 1: Double - The minimum possible distance between any atoms.
RcutCoulombSets a specific radius for each box in the system that short range electrostatic energy will be calculated.
Value 1: Integer - Sets box number (start from ‘0’)
Value 2: Double - The distance to truncate the short rage electrostatic energy at.
Note
The default value for
RcutCoulombis the value ofRcutImportant
In Ewald Summation method, at constant
Toleranceand box volume, increasingRcutCoulombwould result is decreasing reciprocal vector [Fincham 1993]. Decreasing the reciprocal vector decreases the computation time in long range electrostatic calculation.Increasing the
RcutCoulombresults in increasing the computation time in short range electrostatic calculation.Parallelization of Ewald summation method is done on reciprocal vector loop, rather than molecule loop. So, in case of running on multiple CPU threads or GPU, it is better to use the lower value for
RcutCoulomb, to maximize the parallelization efficiency.There is an optimum value for
RcutCoulomb, where result in maximum effeciency of the method. We encourage to run a short simulation with variousRcutCoulombto find the optimum value.
LRCDefines whether or not long range corrections are used.
Value 1: Boolean - True to consider long range correction.
Note
In case of using
SHIFTorSWITCHpotential functions,LRCwill be ignored.ExcludeDefines which pairs of bonded atoms should be excluded from non-bonded interactions.
Value 1: String - Allows you to choose between “1-2”, “1-3”, and “1-4”.
1-2: All interactions pairs of bonded atoms, except the ones that separated with one bond, will be considered and modified using 1-4 parameters defined in parameter file.
1-3: All interaction pairs of bonded atoms, except the ones that separated with one or two bonds, will be considered and modified using 1-4 parameters defined in parameter file.
1-4: All interaction pairs of bonded atoms, except the ones that separated with one, two or three bonds, will be considered using non-bonded parameters defined in parameter file.
Note
The default value for
Excludeis “1-4”.Note
In CHARMM force field, the 1-4 interaction needs to be considered. Choosing “
Exclude1-3” will modify 1-4 interaction based on 1-4 parameters in parameter file. If a kind of force field is used, where 1-4 interaction needs to be ignored, such as TraPPE, either “Exclude1-4” needs to be chosen or 1-4 parameter needs to be assigned to zero in the parameter file.
PotentialDefines the potential function type to calculate non-bonded interaction energy and force between atoms.
Value 1: String - Allows you to pick between “VDW”, “EXP6”, “SHIFT” and “SWITCH”.
VDW: Nonbonded interaction energy and force calculated based on n-6 (Lennard-Johns) equation. This function will be discussed further in the Intermolecular energy and Virial calculation section.
################################# # SIMULATION CONDITION ################################# Temperature 270.00 Potential VDW LRC true Rcut 10 Exclude 1-4
EXP6: Nonbonded interaction energy and force calculated based on exp-6 (Buckingham potential) equation. This function will be discussed further in the Intermolecular energy and Virial calculation section.
################################# # SIMULATION CONDITION ################################# Temperature 270.00 Potential EXP6 LRC true Rcut 10 Exclude 1-4
SHIFT: This option forces the potential energy to be zero at
Rcutdistance. This function will be discussed further in the Intermolecular energy and Virial calculation section.################################# # SIMULATION CONDITION ################################# Temperature 270.00 Potential SHIFT LRC false Rcut 10 Exclude 1-4 RcutCoulomb 0 12.0 RcutCoulomb 1 20.0
SWITCH: This option smoothly forces the potential energy to be zero at
Rcutdistance and starts modifying the potential atRswitchdistance. Depending on force field type, specific potential function will be applied. These functions will be discussed further in the Intermolecular energy and Virial calculation section.
RswitchIn the case of choosing “SWITCH” as potential function, a distance is set in which non-bonded interaction energy is truncated smoothly at
Rcutdistance.Value 1: Double - Define switch distance in angstrom. If the “SWITCH” function is chosen,
Rswitchneeds to be defined; otherwise, the program will be terminated.
################################# # SIMULATION CONDITION ################################# Temperature 270.00 Potential SWITCH LRC false Rcut 12 Rswitch 9 Exclude 1-4
VDWGeometricSigmaUse geometric mean, as required by OPLS force field, to combining Lennard-Jones sigma parameters for different atom types.
Value 1: Boolean - True, uses geometric mean to combine L-J sigmas
Note
The default setting of
VDWGeometricSigmais false to use arithmetic mean when combining Lennard-Jones sigma parameters for different atom types.
ElectroStaticConsiders coulomb interaction or not. This function will be discussed further in the Inter- molecular energy and Virial calculation section.
Value 1: Boolean - True if coulomb interaction needs to be considered and false if not.
Note
To simulate the polar molecule in MARTINI force field,
ElectroStaticneeds to be turn on. MARTINI force field uses short range coulomb interaction with constantDielectric15.0.
EwaldImplements the standard Ewald summation method for electrostatic calculation. This function will be discussed further in the Intermolecular energy and Virial calculation section.
Value 1: Double - True if Ewald summation calculation needs to be considered and false if not.
Note
By default,
ElectroStaticwill be set to true if the Ewald summation method was used to calculate coulomb interaction.
CachedFourierStores the reciprocal terms for Ewald summation calculation to improve code performance. This option increases code performance at the cost of more memory usage. The effects of this option depend on the system size and the value of RcutCoulomb, as the number of k-vectors increases with the value of RcutCoulomb. Small systems may see no or minimal change in performance and memory usage.
Value 1: Boolean - True to store reciprocal terms of Ewald summation calculation for reuse and false to not store them.
Note
The default, the value of
CachedFourieris set to true.Note
The
CachedFourieroption is ignored when running on the GPU.Warning
Monte Carlo moves, such as
MEMC-1,MEMC-2,MEMC-3,IntraMEMC-1,IntraMEMC-2,IntraMEMC-3do not supportCachedFourier.
ToleranceSpecifies the accuracy of the Ewald summation calculation. Ewald separation parameter and number of reciprocal vectors for the Ewald summation are determined based on the accuracy parameter.
Value 1: Double - Sets the accuracy in Ewald summation calculation.
Note
A reasonable value for the accuracy is 0.00001.
If “Ewald” was chosen and no value was set for Tolerance, the program will terminate.
DielectricDefines dielectric constant for coulomb interaction in MARTINI force field.
Value 1: Double - Sets dielectric value used in coulomb interaction.
Note
In MARTINI force field,
Dielectricneeds to be set to 15.0.If MARTINI force field was chosen and
Dielectricwas not specified, a default value of 15.0 will be assigned.
PressureCalcConsiders to calculate the pressure or not. If it is set to true, the frequency of pressure calculation need to be set.
Value 1: Boolean - True enabling pressure calculation during the simulation, false disabling pressure calculation.
Value 2: Ulong - The frequency of calculating the pressure.
1-4scalingDefines constant factor to modify intra-molecule coulomb interaction.
Value 1: Double - A fraction number between 0.0 and 1.0.
Note
CHARMM force field uses a value between 0.0 and 1.0. In MARTINI force field, it needs to be set to 1.0 because 1-4 interaction will not be modified in this force field.
################################# # SIMULATION CONDITION ################################# ElectroStatic true Ewald true Tolerance 0.00001 CachedFourier false 1-4scaling 0.0
RunStepsSets the total number of steps to run (one move is performed for each step) (cycles = this value / number of molecules in the system)
Value 1: Ulong - Total run steps
Note
RunSteps is a delta.
Important
Setting the
RunStepsto zero, and activatingRestartsimulation, will recalculate the energy of stored simulation’s snapshots.EqStepsSets the number of steps necessary to equilibrate the system; averaging will begin at this step.
Value 1: Ulong - Equilibration steps
Note
EqSteps is not a delta. If restarting a simulation with a start step greater than EqSteps, no equilibration is performed.
Note
In GCMC simulation, the
Histogramfiles will be outputed atEqSteps.AdjStepsSets the number of steps per adjustment of the parameter associated with each move (e.g. maximum translate distance, maximum rotation, maximum volume exchange, etc.)
Value 1: Ulong - Number of steps per move adjustment
InitStepSets the first step of the simulation.
Value 1: Ulong - Number of first step of simulation.
Note
Hybrid Monte-Carlo Molecular Dynamics (py-MCMD) requires resetting start step to 0 for combination of NAMD and GOMC data.
################################# # STEPS ################################# RunSteps 25000000 EqSteps 5000000 AdjSteps 1000 InitStep 0
ChemPotFor Grand Canonical (GC) ensemble runs only: Chemical potential at which simulation is run.
Value 1: String - The residue name to apply this chemical potential.
Value 2: Double - The chemical potential value in degrees Kelvin (should be negative).
Note
For binary systems, include multiple copies of the tag (one per residue kind).
If there is a molecule kind that cannot be transfer between boxes (in PDB file the beta value is set to 1.00 or 2.00), an arbitrary value (e.g. 0.00) can be assigned to the residue name.
################################# # Mol. Name Chem. Pot. (K) ################################# ChemPot ISB -968
FugacityFor Grand Canonical (GC) ensemble runs only: Fugacity at which simulation is run.
Value 1: String - The residue to apply this fugacity.
Value 2: Double - The fugacity value in bar.
Note
For binary systems, include multiple copies of the tag (one per residue kind).
If there is a molecule kind that cannot be transfer between boxes (in PDB file the beta value is set to 1.00 or 2.00) an arbitrary value e.g. 0.00 can be assigned to the residue name.
################################# # Mol. Name Fugacity (bar) ################################# Fugacity ISB 10.0 Fugacity Si 0.0 Fugacity O 0.0
DisFreqFractional percentage at which displacement move will occur.
Value 1: Double - % Displacement
RotFreqFractional percentage at which rigid rotation move will occur.
Value 1: Double - % Rotatation
IntraSwapFreqFractional percentage at which molecule will be removed from a box and inserted into the same box using coupled-decoupled configurational-bias algorithm.
Value 1: Double - % Intra molecule swap
Note
The default value for
IntraSwapFreqis 0.000IntraTargetedSwapFreqFractional percentage at which molecule will be removed from the box and inserted into a subvolume within the same box, or deleted from the subvolume and inserted into the same box using coupled-decoupled configurational-bias algorithm.
Value 1: Double - % Intra molecule swap
Note
The default value for
IntraTargetedSwapFreqis 0.000RegrowthFreqFractional percentage at which part of the molecule will be deleted and then regrown using coupled-decoupled configurational-bias algorithm.
Value 1: Double - % Molecular growth
Note
The default value for
RegrowthFreqis 0.000CrankShaftFreqFractional percentage at which crankshaft move will occur. In this move, two atoms that are forming angle or dihedral are selected randomely and form a shaft. Then any atoms or group that are within these two selected atoms, will rotate around the shaft to sample intramolecular degree of freedom.
Value 1: Double - % Crankshaft
Note
The default value for
CrankShaftFreqis 0.000MultiParticleFreqFractional percentage at which multi-particle move will occur. In this move, all molecules in the selected simulation box will be rigidly rotated or displaced simultaneously, along the calculated torque or force, respectively.
Value 1: Double - % Multiparticle
Note
The default value for
MultiParticleFreqis 0.000MultiParticleBrownianFreqFractional percentage at which multi-particle brownian move will occur. In this move, all molecules in the selected simulation box will be rigidly rotated or displaced simultaneously, along the calculated torque or force, respectively.
Value 1: Double - % Multiparticle
Note
The default value for
MultiParticleBrownianFreqis 0.000IntraMEMC-1FreqFractional percentage at which specified number of small molecule kind will be exchanged with a specified large molecule kind in defined sub-volume within same simulation box.
Value 1: Double - % Molecular exchange
Note
IntraMEMC-2FreqFractional percentage at which specified number of small molecule kind will be exchanged with a specified large molecule kind in defined sub-volume within same simulation box. Backbone of small and large molecule kind will be used to insert the large molecule more efficiently.
Value 1: Double - % Molecular exchange
Note
The default value for
IntraMEMC-2Freqis 0.000This move need additional information such as
ExchangeVolumeDim,ExchangeRatio,ExchangeSmallKind,ExchangeLargeKind,SmallKindBackBone, andLargeKindBackBone, which will be explained later.For more information about this move, please refere to MEMC-GCMC and MEMC-GEMC papers.
IntraMEMC-3FreqFractional percentage at which specified number of small molecule kind will be exchanged with a specified large molecule kind in defined sub-volume within same simulation box. Specified atom of the large molecule kind will be used to insert the large molecule using coupled-decoupled configurational-bias.
Value 1: Double - % Molecular exchange
Note
The default value for
IntraMEMC-3Freqis 0.000This move need additional information such as
ExchangeVolumeDim,ExchangeRatio,ExchangeSmallKind,ExchangeLargeKind, andLargeKindBackBone, which will be explained later.For more information about this move, please refere to MEMC-GCMC and MEMC-GEMC papers.
MEMC-1FreqFor Gibbs and Grand Canonical (GC) ensemble runs only: Fractional percentage at which specified number of small molecule kind will be exchanged with a specified large molecule kind in defined sub-volume in dense simulation box.
Value 1: Double - % Molecular exchange
Note
MEMC-2FreqFor Gibbs and Grand Canonical (GC) ensemble runs only: Fractional percentage at which specified number of small molecule kind will be exchanged with a specified large molecule kind in defined sub-volume in dense simulation box. Backbone of small and large molecule kind will be used to insert the large molecule more efficiently.
Value 1: Double - % Molecular exchange
Note
The default value for
MEMC-2Freqis 0.000This move need additional information such as
ExchangeVolumeDim,ExchangeRatio,ExchangeSmallKind,ExchangeLargeKind,SmallKindBackBone, andLargeKindBackBone, which will be explained later.For more information about this move, please refere to MEMC-GCMC and MEMC-GEMC papers.
MEMC-3FreqFor Gibbs and Grand Canonical (GC) ensemble runs only: Fractional percentage at which specified number of small molecule kind will be exchanged with a specified large molecule kind in defined sub-volume in dense simulation box. Specified atom of the large molecule kind will be used to insert the large molecule using coupled-decoupled configurational-bias.
Value 1: Double - % Molecular exchange
Note
The default value for
MEMC-3Freqis 0.000This move need additional information such as
ExchangeVolumeDim,ExchangeRatio,ExchangeSmallKind,ExchangeLargeKind, andLargeKindBackBone, which will be explained later.For more information about this move, please refere to MEMC-GCMC and MEMC-GEMC papers.
SwapFreqFor Gibbs and Grand Canonical (GC) ensemble runs only: Fractional percentage at which molecule swap move will occur using coupled-decoupled configurational-bias.
Value 1: Double - % Molecule swaps
TargetedSwapFreqFor Gibbs and Grand Canonical (GC) ensemble runs only: Fractional percentage at which targeted molecule swap move will occur using coupled-decoupled configurational-bias in the sub-volumes specified.
Value 1: Double - % Molecule targeted swaps
VolFreqFor isobaric-isothermal ensemble and Gibbs ensemble runs only: Fractional percentage at which molecule will be removed from one box and inserted into the other box using configurational bias algorithm.
Value 1: Double - % Volume swaps
#################################
# MOVE FREQEUNCY
#################################
DisFreq 0.39
RotFreq 0.10
IntraSwapFreq 0.10
RegrowthFreq 0.10
CrankShaftFreq 0.10
SwapFreq 0.20
VolFreq 0.01
Warning
All move percentages should add up to 1.0; otherwise, the program will terminate.
ExchangeVolumeDimTo use all variation of
MEMCandIntraMEMCMonte Carlo moves, the exchange sub-volume must be defined. The exchange sub-volume is defined as an orthogonal box with x-, y-, and z-dimensions, where small molecule/molecules kind will be selected from to be exchanged with a large molecule kind.Value 1: Double - X dimension in \(Å\)
Value 2: Double - Y dimension in \(Å\)
Value 3: Double - Z dimension in \(Å\)
Note
Currently, the X and Y dimension cannot be set independently (X = Y = max(X, Y))
A heuristic for setting good values of the x-, y-, and z-dimensions is to use the geometric size of the large molecule plus 1-2 Å in each dimension.
In case of exchanging 1 small molecule kind with 1 large molecule kind in
IntraMEMC-2,IntraMEMC-3,MEMC-2,MEMC-3Monte Carlo moves, the sub-volume dimension has no effect on acceptance rate.
ExchangeSmallKindTo use all variation of
MEMCandIntraMEMCMonte Carlo moves, the small molecule kind to be exchanged with a large molecule kind must be defined. Multiple small molecule kind can be specified.Value 1: String - Small molecule kind (resname) to be exchanged.
ExchangeLargeKindTo use all variation of
MEMCandIntraMEMCMonte Carlo moves, the large molecule kind to be exchanged with small molecule kind must be defined. Multiple large molecule kind can be specified.Value 1: String - Large molecule kind (resname) to be exchanged.
ExchangeRatioTo use all variation of
MEMCandIntraMEMCMonte Carlo moves, the exchange ratio must be defined. The exchange ratio defines how many small molecule will be exchanged with 1 large molecule. For each large-small molecule pairs, one exchange ratio must be defined.Value 1: Integer - Ratio of exchanging small molecule/molecules with 1 large molecule.
LargeKindBackBoneTo use
MEMC-2,MEMC-3,IntraMEMC-2, andIntraMEMC-3Monte Carlo moves, the large molecule backbone must be defined. The backbone of the molecule is defined as a vector that connects two atoms belong to the large molecule. The large molecule backbone will be used to align the sub-volume inMEMC-2andIntraMEMC-2moves, while inMEMC-3andIntraMEMC-3moves, it uses the atom name to start growing the large molecule using coupled-decoupled configurational-bias. For each large-small molecule pairs, two atom names must be defined.Value 1: String - Atom name 1 belong to the large molecule’s backbone
Value 2: String - Atom name 2 belong to the large molecule’s backbone
Important
In
MEMC-3andIntraMEMC-3Monte Carlo moves, both atom names must be same, otherwise program will be terminated.If large molecule has only one atom (mono atomic molecules), same atom name must be used for
Value 1andValue 2of theLargeKindBackBone.
SmallKindBackBoneTo use
MEMC-2, andIntraMEMC-2Monte Carlo moves, the small molecule backbone must be defined. The backbone of the molecule is defined as a vector that connects two atoms belong to the small molecule and will be used to align the sub-volume. For each large-small molecule pairs, two atom names must be defined.Value 1: String - Atom name 1 belong to the small molecule’s backbone
Value 2: String - Atom name 2 belong to the small molecule’s backbone
Important
If small molecule has only one atom (mono atomic molecules), same atom name must be used for
Value 1andValue 2of theSmallKindBackBone.
Here is the example of MEMC-2 Monte Carlo moves, where 7 large-small molecule pairs are defined with an exchange ratio of 1:1: (ethane, methane), (propane, ethane), (n-butane, propane), (n-pentane, nbutane), (n-hexane, n-pentane), (n-heptane, n-hexane), and (noctane, n-heptane).
######################################################################
# MEMC PARAMETER
######################################################################
ExchangeVolumeDim 1.0 1.0 1.0
ExchangeRatio 1 1 1 1 1 1 1
ExchangeLargeKind C8P C7P C6P C5P C4P C3P C2P
ExchangeSmallKind C7P C6P C5P C4P C3P C2P C1P
LargeKindBackBone C1 C8 C1 C7 C1 C6 C1 C5 C1 C4 C1 C3 C1 C2
SmallKindBackBone C1 C7 C1 C6 C1 C5 C1 C4 C1 C3 C1 C2 C1 C1
Here is the example of MEMC-3 Monte Carlo moves, where 7 large-small molecule pairs are defined with an exchange ratio of 1:1: (ethane, methane), (propane, ethane), (n-butane, propane), (n-pentane, nbutane), (n-hexane, n-pentane), (n-heptane, n-hexane), and (noctane, n-heptane).
######################################################################
# MEMC PARAMETER
######################################################################
ExchangeVolumeDim 1.0 1.0 1.0
ExchangeRatio 1 1 1 1 1 1 1
ExchangeLargeKind C8P C7P C6P C5P C4P C3P C2P
ExchangeSmallKind C7P C6P C5P C4P C3P C2P C1P
LargeKindBackBone C4 C4 C4 C4 C3 C3 C3 C3 C2 C2 C2 C2 C1 C1
SmallKindBackBone C1 C7 C1 C6 C1 C5 C1 C4 C1 C3 C1 C2 C1 C1
Here is the example of MEMC-2 Monte Carlo moves, where 1 large-small molecule pair is defined with an exchange ratio of 1:2: (xenon, methane).
######################################################################
# MEMC PARAMETER
######################################################################
ExchangeVolumeDim 5.0 5.0 5.0
ExchangeRatio 2
ExchangeLargeKind XE
ExchangeSmallKind C1P
LargeKindBackBone Xe Xe
SmallKindBackBone C1 C1
SubVolumeBoxDefine which box the subvolume occupies. - Value 1: Integer - Sub-volume id. - Value 2: Integer - Sets box number (first box is box ‘0’).
SubVolumeCenterDefine the center of the static subvolume. - Value 1: Integer - Sub-volume id. - Value 2: Double - x value of SubVolumeCenter \(Å\). - Value 3: Double - y value of SubVolumeCenter \(Å\). - Value 4: Double - z value of SubVolumeCenter \(Å\).
SubVolumePBCDefine which dimensions periodic box wrapping is applied in the subvolume. - Value 1: Integer - Sub-volume id. - Value 2: String - X, Y, Z, XY, XZ, YZ, XYZ (Axes should have no spaced between them)
SubVolumeCenterListDefine the center of the dynamic subvolume by defining the atoms to use for the geometric mean calculation. - Value 1: Integer - Sub-volume id. - Value 2: Integer Range - Atom indices used to calculate geometric center of subvolume.
SubVolumeDimDefine the dimensions of the subvolume. - Value 1: Integer - Sub-volume id. - Value 2: Double - x value of SubVolumeDim \(Å\). - Value 3: Double - y value of SubVolumeDim \(Å\). - Value 4: Double - z value of SubVolumeDim \(Å\).
SubVolumeResidueKindDefine which residue kinds can be inserted or deleted from the subvolume. - Value 1: Integer - Sub-volume id. - Value 2: String - Residue kind inserted/deleted from subvolume - Value .: String - Residue kind inserted/deleted from subvolume - Value .: String - Residue kind inserted/deleted from subvolume - Value N: String - Residue kind inserted/deleted from subvolume
SubVolumeRigidSwapDefine whether molecules are held rigid or the geometry is sampled per the coupled-decoupled CBMC scheme. - Value 1: Integer - Sub-volume id. - Value 2: Boolean - If true the molecule is held rigid. If false, geometry is sampled when inserting in the subvolume.
SubVolumeChemPotDefine the chemical potential of a residue kind in the subvolume. Only used in TargetedSwap, not IntraTargetedSwap. - Value 1: Integer - Sub-volume id. - Value 2: String - Residue kind - Value 3: Double - Chemical potential
SubVolumeFugacityDefine the fugacity of a residue kind in the subvolume. Only used in TargetedSwap, not IntraTargetedSwap. - Value 1: Integer - Sub-volume id. - Value 2: String - Residue kind - Value 3: Double - Chemical potential
######################################################################
# TARGETED SWAP (Static subVolume)
######################################################################
SubVolumeBox 1 0
SubVolumeCenter 1 25.0 25.0 25.0
SubVolumeDim 1 35 35 5
SubVolumeResidueKind 1 TIP3
SubVolumeRigidSwap 1 false
SubVolumeChemPot 1 TIP3 -800
######################################################################
# TARGETED SWAP (Dynamic subVolume)
######################################################################
SubVolumeBox 1 0
SubVolumeCenterList 1 1-402
SubVolumeDim 1 35 35 5
SubVolumeResidueKind 1 TIP3
SubVolumeRigidSwap 1 false
SubVolumeChemPot 1 TIP3 -800
useConstantAreaFor Isobaric-Isothermal ensemble and Gibbs ensemble runs only: Considers to change the volume of the simulation box by fixing the cross-sectional area (x-y plane).
Value 1: Boolean - If true volume will change only in z axis, If false volume will change with constant axis ratio.
Note
By default,
useConstantAreawill be set to false if no value was set. It means, the volume of the box will change in a way to maintain the constant axis ratio.FixVolBox0For adsorption simulation in NPT Gibbs ensemble runs only: Changing the volume of fluid phase (Box 1) to maintain the constant imposed pressure and temperature, while keeping the volume of adsorbed phase (Box 0) fix.
Value 1: Boolean - If true volume of adsorbed phase will remain constant, If false volume of adsorbed phase will change.
CellBasisVectorDefines the shape and size of the simulation periodic cell.
CellBasisVector1,CellBasisVector2,CellBasisVector3represent the cell basis vector \(a,b,c\), respectively. This tag may occur multiple times. It occurs once for NVT and NPT, but twice for Gibbs ensemble or GC ensemble.Value 1: Integer - Sets box number (first box is box ‘0’).
Value 2: Double - x value of cell basis vector \(Å\).
Value 3: Double - y value of cell basis vector \(Å\).
Value 4: Double - z value of cell basis vector \(Å\).
Note
If the number of defined boxes were not compatible to simulation type, the program will be terminated.
Example for NVT and NPT ensemble. In this example, each vector is perpendicular to the other two (\(\alpha = 90, \beta = 90, \gamma = 90\)), as indicated by a single x, y, or z value being specified by each and making a rectangular 3-D box:
############################################ # BOX DIMENSION #, X, Y, Z ############################################ CellBasisVector1 0 40.00 00.00 00.00 CellBasisVector2 0 00.00 40.00 00.00 CellBasisVector3 0 00.00 00.00 80.00
Example for Gibbs ensemble and GC ensemble ensemble. In this example, In the first box, only vector \(a\) and \(c\) are perpendicular to each other (\(\alpha = 90, \beta = 90, \gamma = 120\)), and making a non-orthogonal simulation cell with the cell length \(a = 36.91 Å, b = 36.91 Å, c = 76.98 Å\). In the second box, each vector is perpendicular to the other two (\(\alpha = 90, \beta = 90, \gamma = 90\)), as indicated by a single x, y, or z value being specified by each and making a cubic box:
############################################ # BOX DIMENSION #, X, Y, Z ############################################ CellBasisVector1 0 36.91 00.00 00.00 CellBasisVector2 0 -18.45 31.96 00.00 CellBasisVector3 0 00.00 00.00 76.98 CellBasisVector1 1 60.00 00.00 00.00 CellBasisVector2 1 00.00 60.00 00.00 CellBasisVector3 1 00.00 00.00 60.00
Warning
If
Restartwas activated, box dimension does not need to be specified. If it is specified, program will read it but it will be ignored and replaced by the printed cell dimensions and angles in the restart PDB output file from GOMC (OutputName_BOX_0_restart.pdb andOutputName_BOX_1_restart.pdb).CBMC_FirstNumber of CD-CBMC trials to choose the first atom position (Lennard-Jones trials for first seed growth).
Value 1: Integer - Number of initial insertion sites to try.
CBMC_NthNumber of CD-CBMC trials to choose the later atom positions (Lennard-Jones trials for first seed growth).
Value 1: Integer - Number of LJ trials for growing later atom positions.
CBMC_AngNumber of CD-CBMC bending angle trials to perform for geometry (per the coupled-decoupled CBMC scheme).
Value 1: Integer - Number of trials per angle.
CBMC_DihNumber of CD-CBMC dihedral angle trials to perform for geometry (per the coupled-decoupled CBMC scheme).
Value 1: Integer - Number of trials per dihedral.
################################# # CBMC TRIALS ################################# CBMC_First 10 CBMC_Nth 4 CBMC_Ang 100 CBMC_Dih 30
Next section specifies the parameters that will be used for free energy calculation in NVT and NPT ensembles.
FreeEnergyCalcFor NVT and NPT ensemble only: Considers to calculate the free energy data (the energy different between current lambda state and all other neighboring lambda states, and calculate the derivative of energy with respective to current lambda) or not. If it is set to true, the frequency of free energy calculation need to be set. The free energy data will be printed into Free_Energy_BOX_0_
OutputName.dat.Value 1: Boolean - True enabling free energy calculation during the simulation, false disabling the calculation.
Value 2: Ulong - The frequency of calculating the free energy.
MoleculeTypeSets the solute molecule kind (residue name) and molecule number (residue ID), which absolute solvation free will be calculated for.
Value 1: String - The solute name (residue name).
Value 2: Integer - The solute molecule number (residue ID).
InitialStateSets the index of the
LambdaCoulombandLambdaVDWvectors, to determine the simulation lambda value for VDW and Coulomb interactions.Value 1: Integer - The index of
LambdaCoulombandLambdaVDWvectors.
Note
Multiple initial states need to be run to perform a free energy analysis on the system.
LambdaVDWSets the intermediate lambda states to which solute-solvent VDW interaction to be scaled.
Value 1: Double - Lambda values for VDW interaction in ascending order.
Warning
All lambda values must be stated in the ascending order, otherwise the program will terminate.
LambdaCoulombSets the intermediate lambda states to which solute-solvent Coulombic interaction to be scaled.
Value 1: Double - Lambda values for Coulombic interaction in ascending order.
Warning
All lambda values must be stated in the ascending order, otherwise the program will terminate.
Note
By default, the lambda values for Coulombic interaction will be set to zero if
ElectroStaticorEwaldis deactivated.By default, the lambda values for Coulombic interaction will be set to Lambda values for VDW interaction if
ElectroStaticorEwaldis activated.
-The LambdaVDW and LambdaCoulomb lists must be equal in length.
ScaleCoulombDetermines to scale the Coulombic interaction non-linearly (soft-core scheme) or not.
Value 1: Boolean - True if coulombic interaction needs to be scaled non-linearly, False if coulombic interaction needs to be scaled linearly.
Note
By default, the
ScaleCoulombwill be set to false.ScalePowerSets the \(p\) value in soft-core scaling scheme, where the distance between solute and solvent is scaled non-linearly.
Value 1: Integer - The \(p\) value in the soft-core scaling scheme.
Note
By default, the
ScalePowerwill be set to 2.ScaleAlphaSets the \(\alpha\) value in soft-core scaling scheme, where the distance between solute and solvent is scaled non-linearly.
Value 1: Double - \(\alpha\) value in the soft-core scaling scheme.
Note
By default, the
ScaleAlphawill be set to 0.5.MinSigmaSets the minimum \(\sigma\) value in soft-core scaling scheme, where the distance between solute and solvent is scaled non-linearly.
Value 1: Double - Minimum \(\sigma\) value in the soft-core scaling scheme.
Note
By default, the
MinSigmawill be set to 3.0.
Note
Scaling the distance between solute and solvent using soft-core scheme:
\[r_{sc} = \bigg[\alpha {\big(1 - \lambda \big)}^{p}{\sigma}^6 + {r}^6 \bigg]^{\frac{1}{6}}\]Here is the example of solvation free energy of CO2, in intermediate state 3.
#################################
# FREE ENERGY PARAMETERS
#################################
FreeEnergyCalc true 1000
MoleculeType CO2 1
InitialState 3
ScalePower 2
ScaleAlpha 0.5
MinSigma 3.0
ScaleCoulomb false
#states 0 1 2 3 4
LambdaVDW 0.00 0.50 1.00 1.00 1.00
LambdaCoulomb 0.00 0.00 0.00 0.50 1.00
Output Controls
This section contains all the values that control output in the control file. For example, certain variables control the naming of files outputed of the block-averaged thermodynamic variables of interest, the PDB files, etc.
OutputNameUnique name with no space for simulation used to name the block average, PDB, and PSF output files.
Value 1: String - Unique phrase to identify this system.
################################# # OUTPUT FILE NAME ################################# OutputName ISB_T_270_K
CoordinatesFreqControls output of PDB file (coordinates). If PDB outputing was enabled, one file for NVT or NPT and two files for Gibbs ensemble or GC ensemble will be outputed into
OutputName_BOX_n.pdb, where n defines the box number.Value 1: Boolean - “true” enables outputing these files; “false” disables outputing.
Value 2: Ulong - Steps per dump PDB frame. It should be less than or equal to RunSteps. If this keyword could not be found in configuration file, its value will be assigned a default value to dump 10 frames.
Note
DCDFreq should be used unless the low precision and slower PDB trajectory is needed, perhaps beta and occupancy values are desired. The PDB trajectory is much larger and consumes more disk space.
The PDB file contains an entry for every ATOM, in all boxes read. This allows VMD (which requires a constant number of atoms) to properly parse frames, with a bit of help. Atoms that are not currently in a specific box are given the coordinate (0.00, 0.00, 0.00). The occupancy value corresponds to the box a molecule is currently in (e.g. 0.00 for box 0; 1.00 for box 1).
At the beginning of simulation, a merged PSF file will be outputed into
OutputName_merged.psf, in which all boxes will be outputed. It also contains the topology for every molecule in both boxes, corresponding to the merged PDB format. Loading PDB files into merged PSF file in VMD allows the user to visualize and analyze the results.
DCDFreqControls output of DCD file (binary coordinates). If DCD outputing was enabled, one file for NVT or NPT and two files for Gibbs ensemble or GC ensemble will be outputed into
OutputName_BOX_n.dcd, where n defines the box number.Value 1: Boolean - “true” enables outputing these files; “false” disables outputing.
Value 2: Ulong - Steps per dump PDB frame. It should be less than or equal to RunSteps. If this keyword could not be found in configuration file, its value will be assigned a default value to dump 10 frames.
Note
The DCD file contains an entry for every ATOM, in all boxes read. This allows VMD (which requires a constant number of atoms) to properly parse frames, with a bit of help. Atoms that are not currently in a specific box are given the coordinate (0.00, 0.00, 0.00). The occupancy value corresponds to the box a molecule is currently in (e.g. 0.00 for box 0; 1.00 for box 1).
At the beginning of simulation, a merged PSF file will be outputed into
OutputName_merged.psf, in which all boxes will be outputed. It also contains the topology for every molecule in both boxes, corresponding to the merged PDB format. Loading DCD files into merged PSF file in VMD allows the user to visualize and analyze the results.“The DCD files are single precision binary FORTRAN files, so are transportable between computer architectures. The file readers in NAMD and VMD can detect and adapt to the endianness of the machine on which the DCD file was written, and the utility program flipdcd is also provided to reformat these files if needed. The exact format of these files is very ugly but supported by a wide range of analysis and display programs. The timestep is stored in the DCD file in NAMD internal units and must be multiplied by TIMEFACTOR=48.88821 to convert to fs. Positions in DCD files are stored in Å. Velocities in DCD files are stored in NAMD internal units and must be multiplied by PDBVELFACTOR=20.45482706 to convert to Å/ps. Forces in DCD files are stored in kcal/mol/Å.”
source : https://www.ks.uiuc.edu/Research/namd/2.9/ug/node11.html
RestartFreqControls the output of the last state of simulation at a specified step in
PDB files (coordinates)
PSF files (structure)
XSC files (binary box dimensions)
COOR files (binary coordinates)
If provided as input: VEL files (binary velocity)
Value 1: Boolean - “true” enables printing block average; “false” disables it.
Value 2: Ulong - Number of steps per checkpoint output file.
OutputName_BOX_n_restart.*, where n defines the box number. Header part of this file contains important information and will be needed to restart the simulation:Restart PDB files, one file for NVT or NPT and two files for Gibbs ensemble or GC ensemble, will be outputed with the following information. - Simulation cell dimensions and angles.
Maximum amount of displacement (Å), rotation (\(\delta\)), and volume (\(Å^3\)) that used in Displacement, Rotation, and Volume move.
Note
The restart PDB/PSF/COOR/VEL files contains only ATOM that exist in each boxes at specified steps. These box restart files allows the user to load a box into NAMD and run molecular dynamics in Hybrid Monte-Carlo Molecular Dynamics (py-MCMD).
When restarting the GOMC simulation from two restart files, the order of the molecules in the trajectory may differ preventing trajectory concatenation, unless the CHK file is loaded.
Only restart files must be used to begin a GOMC simulation with
Restartsimulation active. The merged psf is NOT a restart file.CoordinatesFreq must be a common multiple of RestartFreq or vice versa.
CheckpointFreqControls the output of the checkpoint file containing values neccessary to continue a simulation as if it never ended.
Value 1: Boolean - “true” enables printing block average; “false” disables it.
Value 2: Ulong - Number of steps per checkpoint output file.
Note
RestartFreq must also be enabled, and CheckpointFreq must equal RestartFreq.
ConsoleFreqControls the output to STDIO (“the console”) of messages such as acceptance statistics, and run timing info. In addition, instantaneously-selected thermodynamic properties will be output to this file.
Value 1: Boolean - “true” enables message printing; “false” disables outputing.
Value 2: Ulong - Number of steps per print. If this keyword could not be found in the configuration file, the value will be assigned by default to dump 1000 output for RunSteps greater than 1000 steps and 100 output for RunSteps less than 1000 steps.
BlockAverageFreqControls the block averages output of selected thermodynamic properties. Block averages are averages of thermodynamic values of interest for chunks of the simulation (for post-processing of averages or std. dev. in those values).
Value 1: Boolean - “true” enables printing block average; “false” disables it.
Value 2: Ulong - Number of steps per block-average output file. If this keyword cannot be found in the configuration file, its value will be assigned a default to dump 100 output.
HistogramFreqControls the histograms. Histograms are a binned listing of observation frequency for a specific thermodynamic variable. In this code, they also control the output of a file containing energy/molecule samples; it only will be used in GC ensemble simulations for histogram reweighting purposes.
Value 1: Boolean - “true” enables printing histogram; “false” disables it.
Value 2: Ulong - Number of steps per histogram output file. If this keyword cannot be found in the configuration file, a value will be assigned by default to dump 1000 output for RunSteps greater than 1000 steps and 100 output for RunSteps less than 1000 steps.
################################# # STATISTICS Enable, Freq. ################################# CoordinatesFreq true 10000000 RestartFreq true 1000000 CheckpointFreq true 1000000 ConsoleFreq true 100000 BlockAverageFreq true 100000 HistogramFreq true 10000
The next section controls the output of the energy/molecule sample file and the distribution file f or molecule counts, commonly referred to as the “histogram” output. This section is only required if Grand Canonical ensemble simulation was used.
DistNameSets short phrase to naming molecule distribution file.
Value 1: String - Short phrase which will be combined with RunNumber and RunLetter to use in the name of the binned histogram for molecule distribution.
HistNameSets short phrase to naming energy sample file.
Value 1: String - Short phrase, which will be combined with RunNumber and RunLetter, to use in the name of the energy/molecule count sample file.
RunNumberSets a number, which is a part of DistName and HistName file name.
Value 1: Uint – Run number to be used in the above file names.
RunLetterSets a letter, which is a part of DistName and HistName file name.
Value 1: Character – Run letter to be used in above file names.
SampleFreqControls histogram sampling frequency.
Value 1: Uint – the number of steps per histogram sample.
################################# # OutHistSettings ################################# DistName dis HistName his RunNumber 5 RunLetter a SampleFreq 200
OutEnergy, OutPressure, OutMolNum, OutDensity, OutVolume, OutSurfaceTensionEnables/Disables for specific kinds of file output for tracked thermodynamic quantities
Value 1: Boolean – “true” enables message output of block averages via this tracked parameter (and in some cases such as entry, components); “false” disables it.
Value 2: Boolean – “true” enables message output of a fluctuation into the console file via this tracked parameter (and in some cases, such as entry, components); “false” disables it.
The keywords are available for the following ensembles
Keyword
NVT
NPT & Gibbs
GC
OutEnergy
\(\checkmark\)
\(\checkmark\)
\(\checkmark\)
OutPressure
\(\checkmark\)
\(\checkmark\)
\(\checkmark\)
OutMolNum
\(\checkmark\)
\(\checkmark\)
OutDensity
\(\checkmark\)
\(\checkmark\)
OutVolume
\(\checkmark\)
\(\checkmark\)
OutSurfaceTension
\(\checkmark\)
Here is an example:
################################# # ENABLE: BLK AVE., FLUC. ################################# OutEnergy true true OutPressure true true OutMolNum true true OutDensity true true OutVolume true true OutSurfaceTension false false
Binary input file types
- Binary representations of the system.
XSC
COOR
VEL
CHK
“NAMD uses a trivial double-precision binary file format for coordinates, velocities, and forces. Due to its high precision this is the default output and restart format. VMD refers to these files as the ``namdbin'' format. The file consists of the atom count as a 32-bit integer followed by all three position or velocity components for each atom as 64-bit double-precision floating point, i.e., NXYZXYZXYZXYZ... where N is a 4-byte int and X, Y, and Z are 8-byte doubles. If the number of atoms the file contains is known then the atom count can be used to determine endianness. The file readers in NAMD and VMD can detect and adapt to the endianness of the machine on which the binary file was written, and the utility program flipbinpdb is also provided to reformat these files if needed. Positions in NAMD binary files are stored in Å. Velocities in NAMD binary files are stored in NAMD internal units and must be multiplied by PDBVELFACTOR=20.45482706 to convert to Å/ps. Forces in NAMD binary files are stored in kcal/mol/Å.”
XSC (eXtended System Configuration file) File
GOMC allows the box dimensions to be defined in one of three ways:
In the control file
In the header of restart PDB file
In a binary XSC file
The XSC file contains the first step of the simulation, cell vectors, and cell origin. Currently, GOMC only uses the cell vectors.
COOR (binary coordinates) File
GOMC allows the box coordinates to be overwritten by a binary coordinates file. The COOR file should have the same number of atoms in it as the PDB file which it is overwriting. The actual coordinates can vary dramatically, which allows the user to sample the coordinates with other engines (MCMD), or transform it however one sees fit.
VEL (binary velocity) File
GOMC allows the velocities associated with each atom to be maintained and output for continuing MD simulations. In the event a molecule transfer occurs, all the atoms of the transferred molecule are given new velocities by Langevin dynamics. These VEL files must originate from NAMD, as GOMC will not produce them without first being provided them.
CHK (checkpoint) File
GOMC contains several variables which, if not accounted for, will produce different outputs even if the initial conditions are exactly the same. These variables are contained in the checkpoint file, and allow the user to pick up a GOMC simulation where it left off without altering the course of the simulation. Also, the checkpoint file is essential for MCMD as molecules are treated as distinguishable in molecular dynamics due to the fact that MD is a continuous trajectory through time. The checkpoint file contains the original atom order of the molecules, and coordinates and velocities are loaded into this order to ensure the trajectories are consistently arranged.
Checkpoint file contents:
Last simulation step that saved into checkpoint file (Start step can be overriden).
True number of simulation steps that have been run.
Maximum amount of displacement (Å), rotation (\(\delta\)), and volume (\(\AA^3\)) that used in Displacement, Rotation, MultiParticle, and Volume move.
Number of Monte Carlo move trial and acceptance.
Random number sequence.
Molecule lookup object.
Original pdb atoms object to reload new positions into.
Original molecule setup object generated from parsing first PSF files.
Accessory data for coordinating loading the restart coordinates into the original ordering.
If built with MPI and parallel tempering was enabled: Random number sequence for parallel tempering.